
Tokyo Institute of Technology Develops Novel Machine Learning Model for Material Surface Characterization
The design and development of novel materials with superior properties demands a comprehensive analysis of their atomic and electronic structures. Electron energy parameters such as ionization potential (IP), the energy needed to remove an electron from the valence band maximum, and electron affinity (EA), the amount of energy released upon the attachment of an electron to the conduction band minimum, reveal important information about the electronic band structure of surfaces of semiconductors, insulators, and dielectrics. The accurate estimation of IPs and EAs in such nonmetallic materials can indicate their applicability for use as functional surfaces and interfaces in photosensitive equipment and optoelectronic devices.
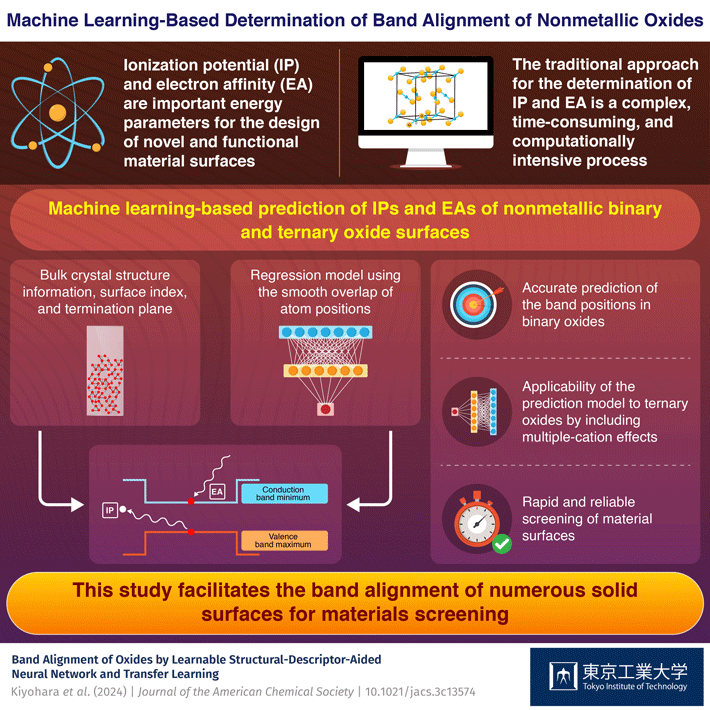
Additionally, IPs and EAs depend significantly on the surface structures, which adds another dimension to the complex procedure of their quantification. Traditional computation of IPs and EAs involves the use of accurate first-principles calculations, where the bulk and surface systems are separately quantified. This time-consuming process prevents quantifying IPs and EAs for many surfaces, which necessitates the use of computationally efficient approaches.
To address the wide-ranging issues affecting the quantification of IPs and EAs of nonmetallic solids, a team of scientists from Tokyo Institute of Technology (Tokyo Tech), led by Professor Fumiyasu Oba, have turned their focus towards machine learning (ML). Their research findings have been published in the Journal of the American Chemical Society![]() .
.
Prof. Oba shares the motivation behind the present research, “In recent years, ML has gained a lot of attention in materials science research. The ability to virtually screen materials based on ML technology is a very efficient way to explore novel materials with superior properties. Also, the ability to train large datasets using accurate theoretical calculations allows for the successful prediction of important surface characteristics and their functional implications.”
The researchers employed an artificial neural network to develop a regression model, incorporating the smooth overlap of atom positions (SOAPs) as numerical input data. Their model accurately and efficiently predicted the IPs and EAs of binary oxide surfaces by using the information on bulk crystal structures and surface termination planes.
Moreover, the ML-based prediction model could ‘transfer learning,’ a scenario where a model developed for a particular purpose can be made to incorporate newer datasets and reapplied for additional tasks. The scientists included the effects of multiple cations in their model by developing ‘learnable’ SOAPs and predicted the IPs and EAs of ternary oxides using transfer learning.
Prof. Oba concludes by saying, “Our model is not restricted to the prediction of surface properties of oxides but can be extended to study other compounds and their properties.”