
Recent Research Enhances Accuracy of Molecular Quantification in High Throughput Sequencing
Accurate sequencing of genetic material is crucial in modern biology, particularly for comprehending and addressing diseases linked to genetic anomalies. However, current methodologies encounter substantial constraints. In a landmark study, an international consortium of researchers, led by Adam Cribbs, Associate Professor in Computational Biology, and Jianfeng Sun, Postdoctoral Research Associate at the Botnar Institute, University of Oxford, have developed an innovative method to correct errors in PCR amplification – a widely used technique used in high-throughput sequencing. By pinpointing PCR artefacts as the primary source of inaccurate quantification, the research, published in Nature Methods, addresses a long-standing challenge in generating accurate absolute counts of RNA molecules, which is crucial for various applications in genomics research.
The researchers focused on Unique Molecular Identifiers (UMIs), which are random oligonucleotide sequences used to remove biases introduced during PCR amplification. While UMIs have been widely adopted in sequencing methods, the study reveals that PCR errors can undermine the accuracy of molecular quantification, particularly across different sequencing platforms.
Jianfeng explained: ‘PCR amplification, essential for most RNA sequencing techniques, can introduce errors, compromising data integrity. We tackled this by synthesising UMI barcodes using homotrimer nucleotide blocks, enhancing error correction and enabling near-absolute RNA molecule quantification, markedly improving molecular counting accuracy.’
Homotrimers are nucleotide sequences consisting of three identical bases, for example AAA, CCC, GGG. By evaluating homotrimers nucleotide similarity, errors are detected and corrected through a “majority vote” method (Figure 1).
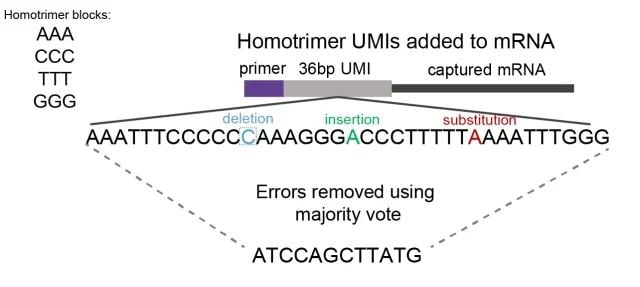
Figure 1: A schematic showing homotrimer UMI majority vote error correction. We constructed UMIs with homotrimeric nucleotide blocks (composed of combinations of AAA, CCC, GGG, TTT). By evaluating trimer nucleotide similarity, errors of deletion, insertion or substitution are identified and corrected through a ‘majority vote’ system, selecting the most frequent nucleotide.
The study demonstrates that homotrimer UMIs significantly outperform traditional monomer UMIs in reducing false positive fold enrichment during the analysis of differentially expressed genes and transcripts (DEGs and DETs). This enhancement is vital for the accurate identification and quantification of DEGs or DETs, particularly in bulk sequencing approaches. Additionally, in single-cell sequencing, where extensive PCR amplification is often required, homotrimer UMIs have proven effective in mitigating the effects of PCR artefacts, thereby substantially improving the reliability of sequencing data.
‘By constructing UMIs from homogenous blocks of nucleosides, we aimed to improve error correction in both short- and long-read sequencing, showcasing our commitment to enhancing sequencing technology applications,’ says Associate Professor Adam Cribbs, senior author of the paper and Group Leader in computational biology.
This research has profound implications. By rectifying PCR errors in UMIs, it greatly boosts molecular quantification accuracy in various sequencing applications. It’s a vital tool for researchers in bulk RNA, single-cell RNA, and DNA sequencing, enabling accurate gene expression and molecular profile analyses. Enhanced UMI error correction not only reduces the incidence of false positives but also offers multiple diagnostic applications, especially in scenarios necessitating longitudinal analysis of samples.